Általánosság
A kifejezés retinitis pigmentosa (RP) azonosítja a genetikai betegségek egy csoportját, amelyet a progresszív retina degeneráció jellemez.

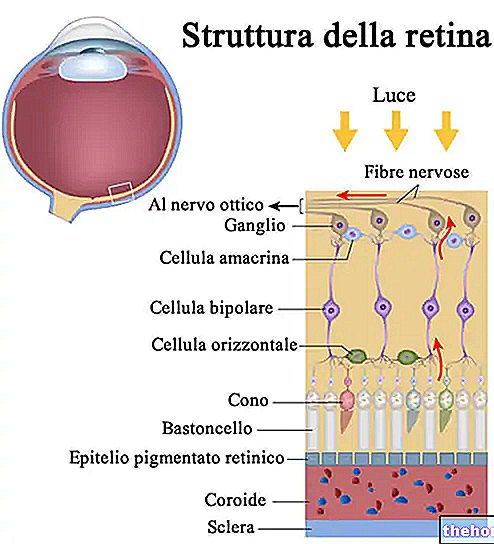
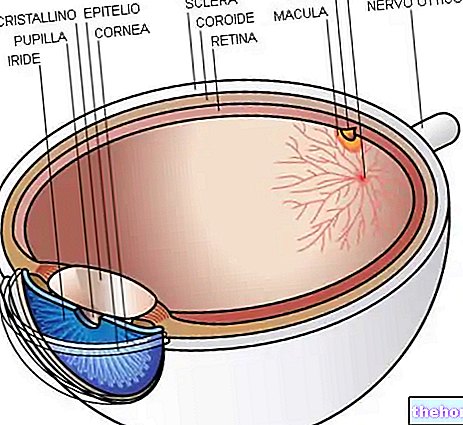
A Retinitis pigmentosa egy retina disztrófia, amelyet a fotoreceptorok fokozatos elvesztése és a pigmenthám diszfunkciója jellemez. Ez azt jelenti, hogy a retina fokozatosan csökkenti annak képességét, hogy vizuális információt továbbítson az agyba a látóidegen keresztül.
A kóros folyamat a retina pigment hámjának megváltozásával kezdődik. A retinitis pigmentosa előrehaladtával elvékonyodnak a retinát ellátó erek, amelyek sorvadnak. A szemfenék vizsgálata után a jellegzetes lerakódások vizuálisan észlelhetők. Retina pigment ( innen ered a betegség neve). Az atrófiás elváltozások és károsodások a látóideget is érinthetik, és fokozatosan a retina fényérzékeny sejtjei elpusztulnak.
A retinitis pigmentosa által érintett betegek kezdetben látási problémákat tapasztalnak, különösen rosszul megvilágított környezetben, és panaszkodnak a perifériás látótér szűkületére. A központi látást a betegség későbbi szakaszaiig kímélik, és a végeredmény drámaian változhat: sok retinitis pigmentosa -ban szenvedő ember látása egész életében korlátozott, míg mások teljesen elveszítik látásukat.
A retinitis pigmentosa örökletes betegség, amelyet főként az egyik vagy mindkét szülő által öröklött genetikai változások okoznak. A genetikai hiba típusa határozza meg, hogy mely retina sejtek vesznek részt leginkább a rendellenességben, és lehetővé teszi klinikai szempontból a különböző állapotok megkülönböztetését. A mai napig több mint 50 különböző genetikai hibát azonosítottak a retinitis pigmentosa -ban. A rendellenességek a szülőkről az utódokra továbbadhatók a három öröklődési minta egyikén keresztül: autoszomális recesszív, autoszomális domináns vagy heteroszomális recesszív (X-kapcsolt vagy X-kapcsolt).
Tünetek
További információ: Retinitis Pigmentosa Tünetek
A retinitis pigmentosa általában serdülőknél és fiatal felnőtteknél fordul elő. A tünetek gyakran 10 és 30 év között jelentkeznek, de a diagnózist kora gyermekkorban vagy sokkal későbbi életkorban állapíthatjuk meg.
A retinitis pigmentosa korai tünetei a következők lehetnek:
- Látási nehézség éjszaka (éjszakai vakság) vagy gyenge fényviszonyok között
- Lassú alkalmazkodás a sötét látástól a fényhez, és fordítva;
- A látótér szűkülése és a perifériás látás elvesztése;
- Fény- és vakító érzékenység.
Néhány tünet az érintett fotoreceptorok típusától függ. A rudak felelősek a fekete -fehér látásért, míg a kúpok lehetővé teszik a színek megkülönböztetését.
A retinitis pigmentosa legtöbb esetben először a rudakat érintik. A gyorsan fejlődő formákban azonban a kúpok is korai stádiumban érintettek lehetnek.
A rudak a retina külső részeiben koncentrálódnak, és a gyenge fény aktiválja őket, így degenerációjuk befolyásolja a perifériás és az éjszakai látást. Kúpok esetén a színérzékelés és a központi látás elvesztése tapasztalható.
Az érintett fotoreceptorok túlsúlyát a páciens genetikai felépítésében meglévő sajátos hiba határozza meg.
Gyakran előfordul, hogy a retinitis pigmentosa első tünete az éjszakai vakság (vagy nocthalopia). Vannak, akik azt tapasztalják, hogy egyre több időre van szükségük ahhoz, hogy alkalmazkodjanak a fényviszonyokhoz, miközben jól megvilágított területről sötétebbre térnek át. A látásvesztés tipikus formája a perifériás látás szűkülését idézi elő (alagút- vagy távcsöves látás); ezt a mintát gyűrűs scotomának nevezik. Néha ez a jelenség hiányozhat a korai szakaszban, de akkor figyelhető meg, ha az egyén gyakran megbotlik tárgyakon, vagy közlekedési balesetbe kerül. nehézségeket tapasztal az olvasásban és a részletes munkában, amely egyetlen tárgyra kell összpontosítania, például egy szál befűzése a tűszembe Sok beteg arról számol be, hogy fényvillanásokat (fotopsziát) lát, amelyeket gyakran apró, villódzó és villogó fényként írnak le.
A betegség előrehaladásának üteme és a látásvesztés mértéke személyenként eltérő. Egyes szélsőséges esetek gyorsan fejlődhetnek két évtizeden belül, mások lassú lefolyással, amely soha nem vezet teljes vaksághoz. A korai megjelenés a retinitis pigmentosa súlyosabb formáiban fordul elő, míg az enyhébb állapotú betegeknél (pl. Autoszomális domináns) az ötödik vagy hatodik évtizedben alakulhat ki a betegség. Az X-kapcsolt retinitis pigmentosa családokban a férfiak gyakrabban érintettek a nőknél súlyosabb; a nők viszont továbbítják a genetikai jellemzőket (az X kromoszómán hordozzák a megváltozott gént), és ritkábban nyilvánulnak meg a rendellenesség tünetei.
Szövődmények
A Retinitis pigmentosa tovább fog fejlődni, bár lassan. A teljes vakság azonban ritka, de a perifériás és központi látás jelentős csökkenése előfordulhat.
A retinitis pigmentosa -ban szenvedő betegeknél korai életkorban gyakran kialakul a retina duzzanata (makulaödéma) vagy szürkehályog. Ezek a szövődmények kezelhetők, ha zavarják a látást.
Kapcsolódó betegségek
Általában a retinitis pigmentosa-ban szenvedő betegnek nincs más rendellenessége, és ebben az esetben "nem szindrómás" vagy egyszerű retinitis pigmentosa-ról beszélünk. Ennek ellenére számos szindrómának van néhány klinikai tünete ezzel a szembetegséggel; a leggyakoribb az Usher-szindróma, amely a retinitis pigmentosa-ban szenvedő betegek körülbelül 10-30% -át érinti, és egyidejű veleszületett vagy progresszív halláskárosodással jár. Leber veleszületett amaurosisában azonban a gyermekek az első hat hónapban megvakulhatnak, vagy majdnem vakká válhatnak, A retinitis pigmentosa-val kapcsolatos egyéb betegségek közé tartozik a Bardet-Biedl-szindróma és a Refsum-kór.
Okoz
A betegséget számos genetikai hiba okozhatja: valójában számos gén létezik, amelyek, ha a változás befolyásolja őket, retinitis pigmentosa fenotípust okozhatnak. Ezek általában a látást lehetővé tevő transzdukciós kaszkádban részt vevő fehérjéket kódolják, a sejtek transzkripcióját (amelyek téves üzeneteket küldenek a retina sejtekhez) vagy a fotoreceptorok szerkezetét alkotó elemekhez. Öröklött génmutációk vannak jelen a sejtekben a fogantatás pillanatától kezdve; gyakori rendellenességek közé tartozik az RP1 gének (retinitis pigmentosa-1, autoszomális domináns) , RHO (RP4, autoszomális domináns) és RDS (RP7, autoszomális domináns). A retinitis pigmentosa nem öröklődő okai ritkák, de lehetőség van egy egyedi eset megtalálására (spontán mutáció), amelyben nincs jelen családtörténet a betegség.


.jpg)











---ena-screening-o-pannello-ena.jpg)