Mi a Marfan -szindróma?
A Marfan -szindróma a kötőszövet összetett örökletes rendellenességét írja le, amely elsősorban a szemet, a szív- és érrendszert, valamint a mozgásszervi rendszert érinti. Tekintettel azonban arra, hogy minden szerv kötőszövetből áll, a Marfan -szindróma ideális esetben elpusztíthatja és erősen zavarhatja bármely anatómiai hely növekedését és működését.
A szindrómát autoszomális domináns tulajdonságként továbbítják: ezért egy súlyos genetikai betegséggel kell szembenéznünk, amelynek rendkívül változó "fenotípusos kifejeződése van (a hibák családonként vagy betegenként nagyon eltérőek lehetnek).
Ami a Marfan-szindrómát kiváltja, az az FBN1 gén megváltozása (a 15. kromoszómán), amely a fibrillin-1-et kódolja, egy nagyon fontos kötő glikoproteint, amely a mikrofibrillumok strukturális támogatását képezi.
Mikroszálak: a fibrillinből álló mikrofibrillumok jelen vannak az extracelluláris mátrixban, amelyben összefonódnak az elasztin lerakódásához a rugalmas szálakban. Bár a testben mindenütt jelen vannak, a mikrofibrillumok mindenekelőtt az aortában, a szalagokban és a zónákban találhatók ciliáris testek (szemszinten).
Mivel ez egy autoszomális domináns betegség, csak azokat a gyermekeket érinti a Marfan-szindróma, akik mindkét szülőtől örökölték a megváltozott FBN-1 gént. Mindazonáltal minden negyedik esetből a betegség spontán mutációk eredménye olyan betegeknél, akiknek nincs családtörténete.
A betegség neve a francia gyermekorvosból származik, aki először írta le 1896 -ban (A. Marfan), utána 1991 -ig kellett várni a tüneti megnyilvánulásban szerepet játszó megváltozott gén azonosítására: a felfedező F. Ramirez volt.
Nézd meg a videót
- Nézd meg a videót a youtube -on
Okoz
Említettük, hogy a Marfan-szindróma a fibrillin-1-et kódoló gén mutációjának azonnali kifejeződése.
A FIBRILLIN 1 az elasztin glikoprotein összetevője, amely elengedhetetlen a szövetek rugalmasságának és szilárdságának biztosításához és fenntartásához. Fiziológiai körülmények között a fibrillin 1 kötődik egy másik fehérjéhez, a TGF-béta (vagy transzformáló növekedési faktor béta) néven. Úgy tűnik, hogy a TGF-béta részt vesz az erek simaizmait és az extracelluláris mátrixot érintő káros folyamatokban. Ezekből a feltételezésekből kiindulva egyes szerzők meg vannak győződve arról, hogy a Marfan-szindróma az FBN-1 gén mutációja mellett a TGF-béta túlzott mennyiségének is köszönhető, különösen az aortában, a szívbillentyűkben és a tüdőben.

Előfordulása
Becslések szerint a Marfan-szindróma minden 3000-5000 születésből 1-et érint, és válogatás nélkül fordul elő férfiak és nők között. A statisztikák azt mutatják, hogy a betegek 75% -a pozitív családtörténettel rendelkezik; a fennmaradó 25% -ban az ok szórványos mutációkban rejlik, amelyek valamilyen módon összefüggésbe hozhatók az apa kora fogantatásának pillanatával.
A Marfan -szindróma rendkívül súlyos formáiban szenvedő gyermekek "várható élettartama kevesebb, mint egy év".
A nyílt szívsebészeti stratégiák kifejlesztése előtt a legtöbb Marfan -szindrómás beteg átlagos élettartama 32 év volt; az orvosi és farmakológiai terápiák folyamatos fejlesztésének köszönhetően a Marfan -szindrómában szenvedők jelenleg átlagosan 60 évig élnek.
jelek és tünetek
További információ: Marfan -szindróma tünetei
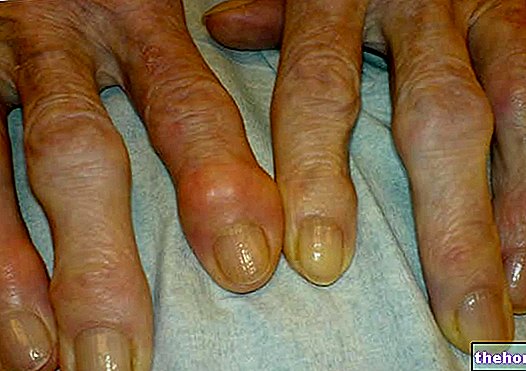
A Marfan -szindróma teljesen tünetmentes. Az érintett betegek túlzottan karcsú szerkezetűek, aránytalanul magasak és vékonyak. Az alsó és felső végtagok sokkal hosszabbak, mint a törzs (dolicostenomegalia). Szó esik arról is arachnodactyly hogy a legjobban kifejezze a Marfan -szindrómában szenvedőkre jellemző túlzott ujjhossz fogalmát: a kezeket ezért egy pók lábához hasonlítják.
A magasság tekintetében ezeknek a betegeknek a termete átlagos, a 97 -es százalék felett van.
A Marfan -szindrómás betegeknél gyakran előforduló egyéb megkülönböztető jellemzők közül emlékszünk még:
- A karok kinyitása nagyobb, mint a magasság
- Laza ízületek → túlzott ízületi mobilitás
- A mellkas deformitása
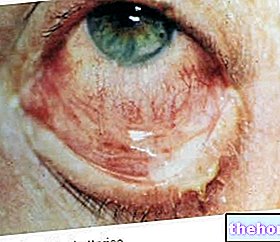
- A lencse elmozdulása
- A felsőtest kevésbé fejlett, mint az alsó rész
- Spontán pneumothorax (11%)
- Gerincferdülés
- Bőr striae a comb, a hát, a deltoid, a mellkas szintjén
A Marfan -szindrómával kapcsolatos legproblémásabb jelek között emlékszünk a szívbillentyű prolapsusára és a mitrális billentyű elégtelenségére: hasonló állapot könnyen kedvezhet az aortagyűrű tágulásának és az aorta boncolásának.
A táblázat a Marfan -szindrómás betegeknél tapasztalható jeleket mutatja. Az ott leírt karakterek nem mindig vannak jelen, de jó részük megtalálható.
Lehetséges tünetek
Bőr
Striák a mellkasi, ágyéki és szakrális területen
Szemek
Látásváltozás, asztigmatizmus, retinaleválás, zárt zugú glaukóma, lencse luxáció, myopia
Csontozat
Arthralgia, kyphoscoliosis, dolichostenomelia (a végtagok túlzott fejlődése a törzshez képest), hipermobilitás, magas szájpadlás, deformált mellkas, lapos lábak, feszes és vékony csukló, a szegycsont kóros visszatérése / kiugrása, gerincferdülés, ívelt vállak, spondylolisthesis
Ujjak
arachnodactyly
Tüdő
Spontán pneumothorax, dyspnoe, idiopátiás obstruktív tüdőbetegség
Arcváltozások
Nyálkahártya -szájpad (a szájpad rendellenessége), mandibularis retrognathia (az állkapocs fejlődési rendellenessége), megnyúlt arc
Szív
Angina pectoris, hasi aorta aneurizma, szívritmuszavar, mellkasi aorta dilatáció / szakadás / boncolás, aorta elégtelenség, mitrális billentyű prolapsus
Nyelv
Nehézség a beszéddel
Diagnózis
Figyelembe véve a több mint 200 lehetséges mutációt, a genetikai markerek használata diagnosztikai célokra szinte lehetetlen.
A Marfan -szindróma értékelése nem mindig ilyen közvetlen, mivel a mutáció fenotípusos kifejeződése nem mindig nyilvánvaló és könnyen azonosítható. A diagnosztikai késleltetés komolyan veszélyeztetheti a beteg túlélését: gondoljunk csak például arra, hogy nem ismerjük fel a szív- és érrendszeri problémákat.
A Marfan -szindróma diagnosztikai kritériumait nemzetközi szinten 1996 -ban állították össze: a diagnózis a "családtörténet vizsgálatából áll, amely a kisebb és nagyobb mutatók a szindrómából.
A számos használt diagnosztikai vizsgálat közül néhány:
- echokardiogram
- mágneses angiorezonancia és CT (az aorta vizsgálatához)
- mágneses rezonancia angiográfia (MRA) kontraszt folyadékkal (az aorta belső szerkezetének kiemelésére)
- réslámpás vizsgálat (a lencse esetleges elmozdulásának elemzésére)
- szemnyomás mérése (a glaukóma lehetséges jelenlétének kiemelésére)
- genetikai vizsgálatok (ajánlott a gyermek fogantatása előtt annak megállapítására, hogy a szindróma vagy sem)
Terápiák
Mivel ez genetikai betegség, nincs olyan gyógyszer vagy kezelés, amely visszafordíthatná a betegséget.
A gyógyszerek alkalmazása azonban elengedhetetlen a tünetek enyhítéséhez és a szövődmények, különösen a szívbetegségek elkerülése érdekében.Ebből a célból különösen alkalmasak a vérnyomáscsökkentő gyógyszerek, például a szartánok (mindenekelőtt), az ACE -gátlók és a béta -blokkolók.
A Marfan -szindróma összefüggésében a gerincferdülésben is szenvedő betegek speciális kezelést, valamint a glaukóma által érintett betegeket követhetnek.
A sebészet elképzelhető a kóros aortatágulat korrigálására, amely elem gyakran egyesíti a Marfan -szindrómás betegek többségét.
Folytatás: Marfan -szindróma - gyógyszerek és kezelés "